Permet de savoir si deux échantillons ou plus sont issus d’une même population ou pour le dire autrement, les groupes créées ont-ils la même moyenne.
L’ANOVA permet d’étudier l’influence d’au moins une variable qualitative ayant deux modalités ou plus, sur une variable quantitative.
D’un point de vue pratique, l’ANOVA cherche à savoir si les moyennes des groupes sont globalement différentes ou pour le dire autrement, si la variation intragroupe est plus faible que la variation intergroupe.
Le principe de l’ANOVA est de décomposer la variabilité totale des données en deux :
_ la variabilité factorielle : la variabilité entre groupes, c’est-à-dire la différence entre la moyenne de toutes les données et les moyennes de chaque groupe (cf. Figure 1).
_ la variabilité résiduelle : la variabilité qui reste une fois que la variabilité due au groupe est retirée c’est-à-dire la différence entre la moyenne du groupe et la valeur de chaque échantillon (cf. Figure 2).
library(tidyverse)
── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
✔ dplyr 1.1.3 ✔ readr 2.1.4
✔ forcats 1.0.0 ✔ stringr 1.5.0
✔ ggplot2 3.4.4 ✔ tibble 3.2.1
✔ lubridate 1.9.3 ✔ tidyr 1.3.0
✔ purrr 1.0.2
── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
✖ dplyr::filter() masks stats::filter()
✖ dplyr::lag() masks stats::lag()
ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
Les données doivent provenir d’un échantillonnage aléatoire et les groupes doivent-être indépendants entre eux.
Normalité des données
Les données au sein de chaque groupe doivent suivre une loi normale ou être approximé par une loi normale (n > 30).
shapiro.test(iris$Sepal.Length)
Shapiro-Wilk normality test
data: iris$Sepal.Length
W = 0.97609, p-value = 0.01018
# les données ne suivent pas une loi normalemap(.x =c(iris$Species |>levels()),.f =~shapiro.test(filter(iris, Species == .x)$Sepal.Length) )
[[1]]
Shapiro-Wilk normality test
data: filter(iris, Species == .x)$Sepal.Length
W = 0.9777, p-value = 0.4595
[[2]]
Shapiro-Wilk normality test
data: filter(iris, Species == .x)$Sepal.Length
W = 0.97784, p-value = 0.4647
[[3]]
Shapiro-Wilk normality test
data: filter(iris, Species == .x)$Sepal.Length
W = 0.97118, p-value = 0.2583
# les données au sein de chaque groupe suivent des lois normales
Homogénéité des variances
Les groupes doivent avoir une variance similaire.
Le test de Bartlett permet de tester la variance de plus de deux groupes.
bartlett.test(Sepal.Length ~ Species, data = iris)
Bartlett test of homogeneity of variances
data: Sepal.Length by Species
Bartlett's K-squared = 16.006, df = 2, p-value = 0.0003345
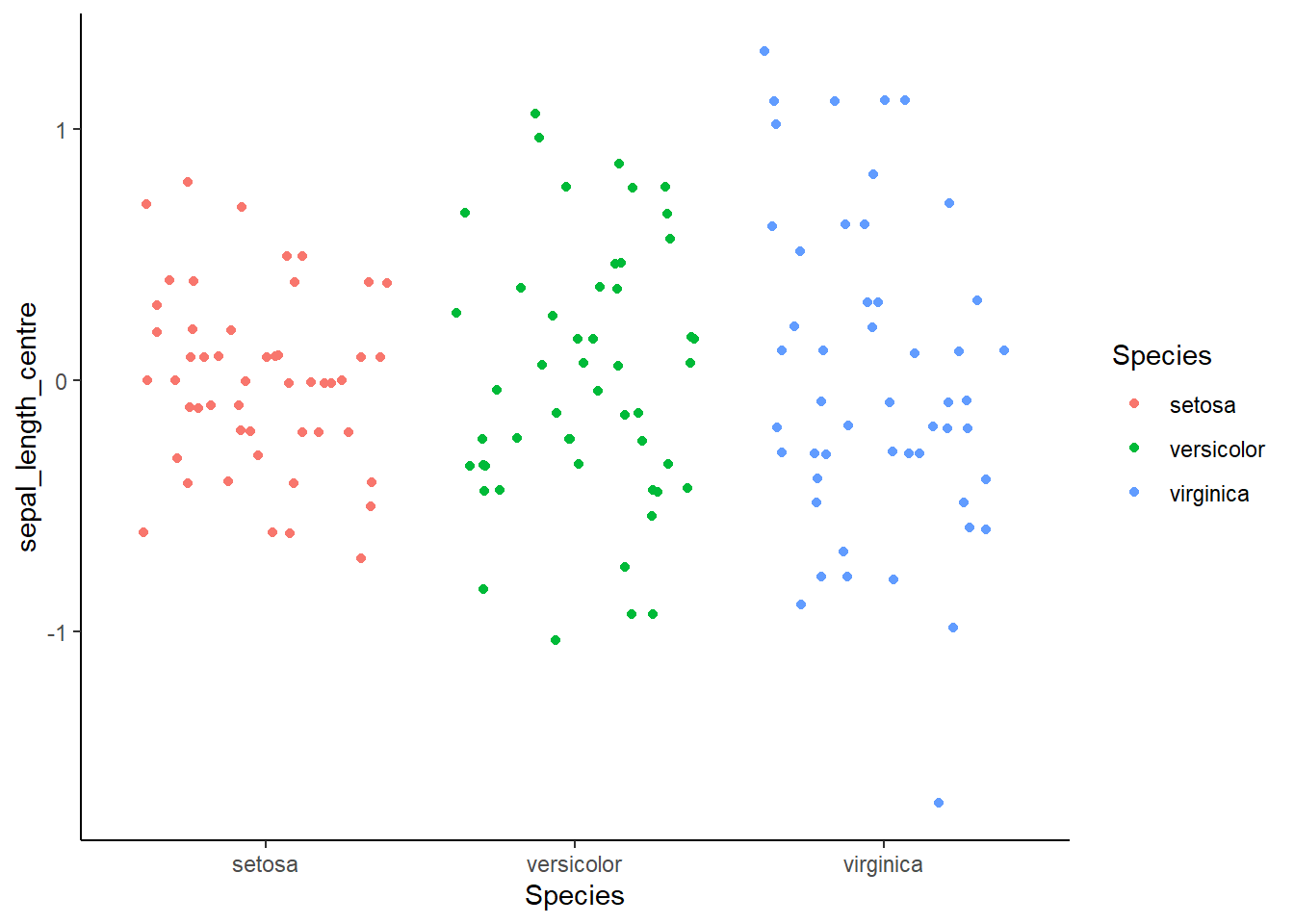
iris |>left_join(iris_moyenne) |>mutate(sepal_length_centre = Sepal.Length - moyenne) |>ggplot() +aes(x = Species, color = Species, y = sepal_length_centre) +geom_jitter() +theme_classic()
Joining with `by = join_by(Species)`
Warning
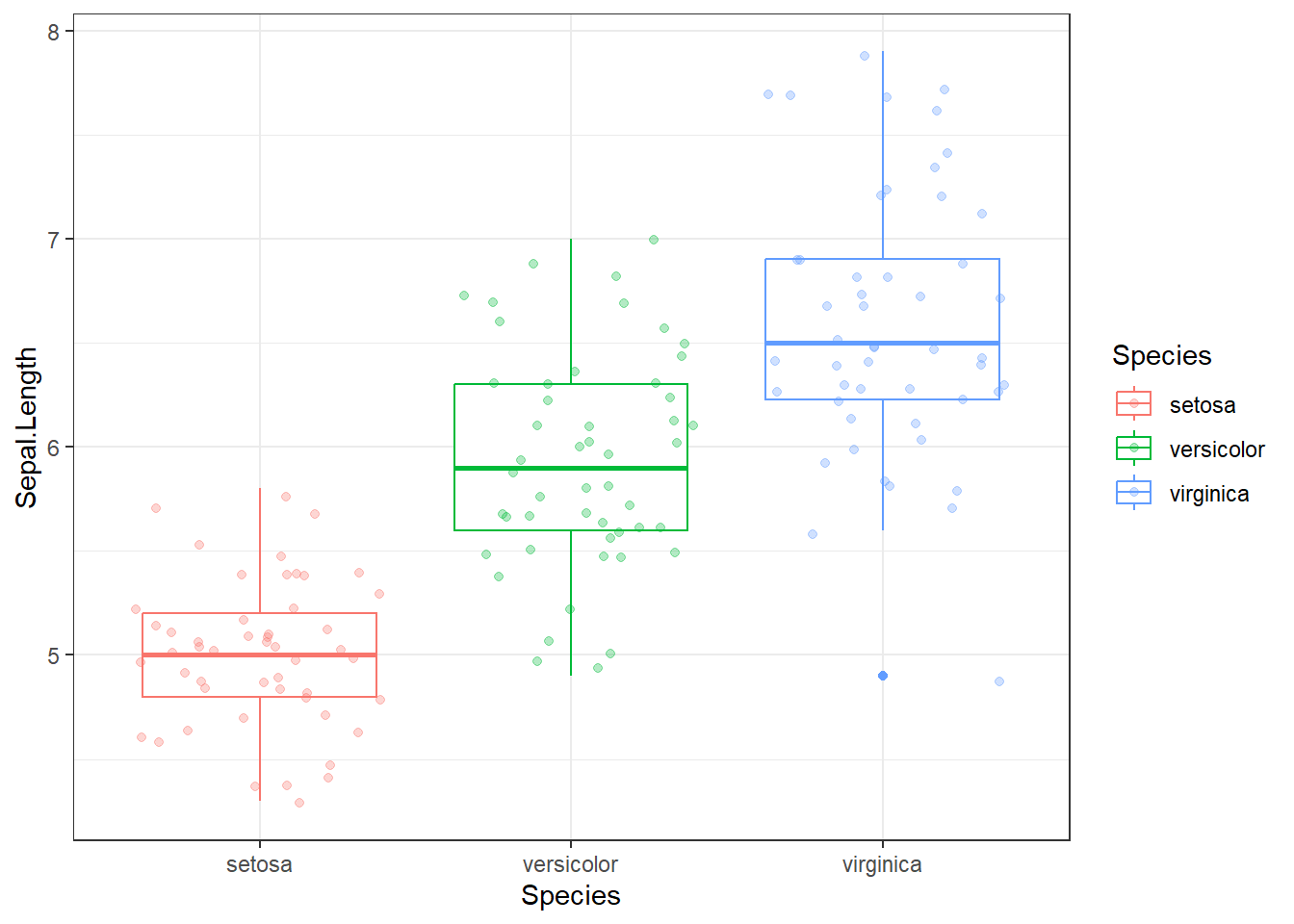
Il ne faut pas faire d’ANOVA ici, les groupes n’ont pas la même variance !
Réalisation d’une ANOVA
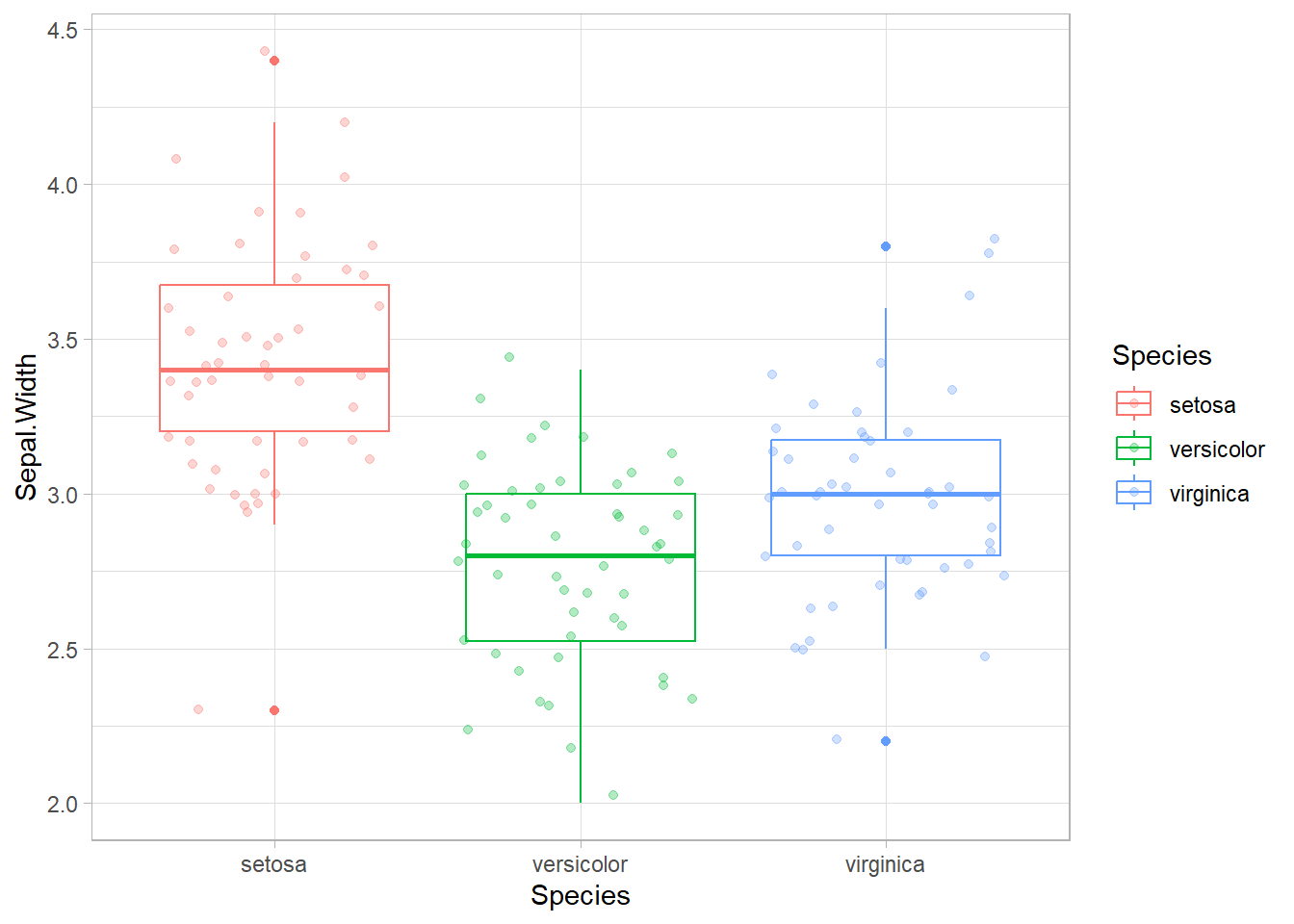
Comme la longueur des sépales ne peut pas être utilisée, on va le faire sur la largeur des sépales.
Vérification des données
map(.x =c(iris$Species |>levels()),.f =~shapiro.test(filter(iris, Species == .x)$Sepal.Width) )
[[1]]
Shapiro-Wilk normality test
data: filter(iris, Species == .x)$Sepal.Width
W = 0.97172, p-value = 0.2715
[[2]]
Shapiro-Wilk normality test
data: filter(iris, Species == .x)$Sepal.Width
W = 0.97413, p-value = 0.338
[[3]]
Shapiro-Wilk normality test
data: filter(iris, Species == .x)$Sepal.Width
W = 0.96739, p-value = 0.1809
bartlett.test(Sepal.Width ~Species, data = iris)
Bartlett test of homogeneity of variances
data: Sepal.Width by Species
Bartlett's K-squared = 2.0911, df = 2, p-value = 0.3515
Les données suivent des lois normales et les variances sont similaires.
Réalisation de l’ANOVA
Important
La détermination d’un modèle ANOVA doit-être réalisé avec la fonction aov() du package {stats}. Les fonctions anova() du package {stats} ou Anova() du package {car} permet de réaliser une analyse de variance/déviance sur un modèle donc par exemple le résultat de aov() mais pas que
anova_sepal_largeur <-aov(Sepal.Width ~ Species, data = iris)summary(anova_sepal_largeur)
Df Sum Sq Mean Sq F value Pr(>F)
Species 2 11.35 5.672 49.16 <2e-16 ***
Residuals 147 16.96 0.115
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
anova(anova_sepal_largeur)
Analysis of Variance Table
Response: Sepal.Width
Df Sum Sq Mean Sq F value Pr(>F)
Species 2 11.345 5.6725 49.16 < 2.2e-16 ***
Residuals 147 16.962 0.1154
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
J’ai oublié de le préciser dans le live sur twitch mais il faut penser à vérifier que les résidus (la différence entre les valeurs prédites et observées) suivent une loi normales et soient homogènes !
shapiro.test(anova_sepal_largeur$residuals)
Shapiro-Wilk normality test
data: anova_sepal_largeur$residuals
W = 0.98948, p-value = 0.323
# A tibble: 3 × 2
Species somme_rang
<fct> <int>
1 setosa 1448
2 versicolor 4115
3 virginica 5762
Une fois que le rang de chaque groupe calculé, la statistique de test va être calculé et comparer à une valeur seuil.
WarningLes limites
_ échantillonnage aléatoire
_ indépendance des groupes
_ Plus de 5 observations par groupe
Utilisation de la fonction kruskal.test() du package {stats}
Comme la longueur des sépales n’avaient pas la même variance en fonction de l’espèce, il n’est pas possible de réaliser une ANOVA.
Le test de Kruskal-Wallis est conseillé ici.
kruskal.test(Sepal.Length ~ Species, data = iris)
Kruskal-Wallis rank sum test
data: Sepal.Length by Species
Kruskal-Wallis chi-squared = 96.937, df = 2, p-value < 2.2e-16
Test-post hoc de Nemenyi
summary( PMCMRplus::kwAllPairsNemenyiTest(data = iris, Sepal.Length ~ Species ))
Warning in kwAllPairsNemenyiTest.default(c(5.1, 4.9, 4.7, 4.6, 5, 5.4, 4.6, :
Ties are present, p-values are not corrected.
Pairwise comparisons using Tukey-Kramer-Nemenyi all-pairs test with Tukey-Dist approximation